
1.はじめに
アトピー性皮膚炎治療のための抗インターロイキン-4受容体(IL-4R)抗体を含有する医薬組成物に関するリジェネロン及びサノフィの特許に対する無効審判請求を不成立とした審決を不服として科研製薬が提起した審決取消訴訟(知財高裁令和5年(行ケ)10019)で、知財高裁は、本件審決の判断に誤りはなく、科研製薬が主張する取消事由(進歩性、サポート要件及び実施可能要件に関する誤り)はいずれも理由がないと判断した。
本件特許は、2023年に世界で116億ドルを売り上げたデュピクセント®(米国販売名はDupixent®)(有効成分:ヒト型抗ヒトIL-4/13受容体モノクローナル抗体であるデュピルマブ)のアトピー性皮膚炎患者における有効性を確認する臨床試験結果に基づく医薬用途発明に係るものであり、日本におけるアトピー性皮膚炎治療に関するデュピクセント®の効能効果を保護するものであると同時に、機能的に表現された抗IL-4受容体抗体のアトピー性皮膚炎治療に関する医薬用途発明をその技術的範囲とするものである。
科研製薬は、ニューマブ社とアトピー性皮膚炎を対象としてIL-4受容体αとIL-31に結合する二重特異性抗体を開発していたことから、この競合関係に今回の特許紛争の火種があったと想像される。
進歩性欠如の無効理由として科研製薬が挙げた引用文献は、本件特許の明細書に実施例としてその結果が記載されている、臨床試験情報データベースClinicalTrials.govに登録されたデュピルマブの臨床試験(フェーズ2)のプロトコル(試験実施計画書)であった。
本稿は、それら臨床試験プロトコルの公開と臨床試験結果に基づく本件特許との関係を整理した上で、本件訴訟における裁判所の判断と欧米での審査状況を紹介しつつ、臨床試験プロトコルの公開が特許性に与える影響から、臨床試験開始(プロトコル公開)前に出願(新規性・進歩性重視)すべきなのか、臨床試験結果が出てから出願(記載要件重視)すべきなのかというジレンマについて、まとまりのない思いついたままの感想を述べるものである。
2.事件の背景
本件訴訟(知財高裁令和5年(行ケ)10019)は、発明の名称を「IL-4Rアンタゴニストを投与することによるアトピー性皮膚炎を処置するための方法」とする発明に関するリジェネロン及びサノフィ(以下「被告ら」)の特許第6353838号(以下「本件特許」)に対して科研製薬(以下「原告」)がした無効審判請求(無効2021-800003号事件)を不成立とした審決の取消訴訟であり、争点(取消事由)は、①進歩性の欠如、②サポート要件違反、③実施可能要件違反である。
本件訂正発明1は以下のとおり。
患者において中等度から重度のアトピー性皮膚炎(AD)を処置する方法に使用するための治療上有効量の抗ヒトインターロイキン-4受容体(IL-4R)抗体またはその抗原結合断片を含む医薬組成物であって、ここで前記患者が局所コルチコステロイドまたは局所カルシニューリン阻害剤による処置に対して十分に応答しないかまたは前記局所処置が勧められない患者である前記医薬組成物。
原審(無効審判)において、原告が主張した無効理由は、甲1に記載された引用発明に基づく進歩性の欠如(無効理由1)、サポート要件違反(無効理由2)及び実施可能要件違反(無効理由3)であった。
甲1(Clinical Trials. Gov archive, History of Changes for Study: NCT 01548404, Study of REGN668 in Adult Patients With Extrinsic Moderate-to-Severe Atopic Dermatitis)は、被告らが、米国食品医薬品局(FDA)に提出した臨床試験のプロトコル(試験実施計画書)(情報データベースからの出力文書)であり(参照URL: https://www.clinicaltrials.gov/study/NCT01548404)、その治験薬組成物(の一部)であるREGN668は、抗IL-4R抗体であり、本件明細書に本件訂正発明1の実施例として記載されている「mAb1」と同一物質であった(注:開発コード名であったREGN668は、その後、一般名としてデュピルマブ(dupilumab)と命名される)。
甲1(臨床試験のプロトコル)には、特定のアトピー性皮膚炎患者を対象に効果を確認する臨床試験(フェーズ2)が実施されている治験薬としてのREGN668(引用発明)が記載されており、本件訂正発明1と引用発明との一致点及び相違点は下記のとおりであると認定された。
「中等度から重度のアトピー性皮膚炎(AD)患者に対する効果、安全性などを評価するための試験に使用される、REGN668を含む治験薬組成物であって、ここで前記患者が18歳以上で、少なくとも3年間の慢性アトピー性皮膚炎を患っており、局所コルチコステロイド又は局所カルシニューリン阻害剤による処置に対して十分に応答しない患者である前記治験薬組成物。」
【一致点】
抗ヒトIL-4R抗体又はその抗原結合断片を含む組成物であって、中等度から重度のアトピー性皮膚炎(AD)であって、局所コルチコステロイド又は局所カルシニューリン阻害剤による処置に対して十分に応答しない患者に投与されるものである点。
【相違点】
本件訂正発明1は、中等度から重度のアトピー性皮膚炎(AD)であって、局所コルチコステロイド又は局所カルシニューリン阻害剤による処置に対して十分に応答しない患者を処置する方法に使用するための、治療上有効な量の抗ヒトIL-4R抗体又はその抗原結合断片を含む医薬組成物であるのに対し、引用発明は、治験薬組成物である点。
「医薬」であるか「治験薬」(臨床効果を試している段階)であるかだけの違い・・・。
特許庁は、原告が主張する進歩性の欠如(無効理由1)について
「乙1によれば、フェーズ2臨床試験の成功の確率は他のどのフェーズよりもはるかに低く(乙1ア)、アレルギー疾患の場合、33%程度であり(乙1イ)、このことからすると、フェーズ2臨床試験が行われていることから直ちに、当該治験薬が試験結果を見るまでもなく当然に治療上有効であると当業者が理解するとはいえない。また、甲2~6を検討しても、本件特許の優先日前に、アトピー性皮膚炎患者に抗ヒトIL-4R抗体が投与されて、実際に治療効果が得られたことを示す証拠はない。」
などと述べ、医薬組成物であるという相違点に係る構成を備え、本件訂正発明1に該当する患者において、実際に本件明細書に示されたアトピー性皮膚炎の臨床症状の改善効果を示すものとすることは、甲1等の記載から当業者が容易になし得たことであるとはいえない(進歩性あり)と判断した。
また、サポート要件違反(無効理由2)及び実施可能要件違反(無効理由3)によっても無効とされるべきものではないと判断した。
原告は、特許庁が原告による無効審判の請求は成り立たないとの審決(以下「本件審決」)をしたため、その取消しを求める審決取消訴訟(本件訴訟)を提起した。
3.裁判所の判断
知財高裁・第4部(以下「裁判所」)は、原告主張の取消事由はいずれも認められないとして、原告の請求を棄却した。
1 原告の請求を棄却する。
2 訴訟費用は原告の負担とする。
以下に、本件訂正発明1についての裁判所の判断を紹介する。
(以下、判決文において、抗ヒトインターロイキン-4受容体(IL-4R)抗体を「本件抗体」と、本件抗体及びその抗原結合断片を併せて「本件抗体等」と、請求項1記載の対象患者を「本件患者」ということがある。)
(1)取消事由1(進歩性判断の誤り)について
容易想到性の判断の誤りについて、原告は、
「甲1の試験(第Ⅱ相試験)に先立ってアトピー性皮膚炎患者に対するREGN668の第Ⅰ相試験が行われ、引用発明に係るREGN668が医薬品としての有用性が期待できる薬物であると既に判断されており、アトピー性皮膚炎がTh2/IL-4等が優勢な疾患であるという正しい技術常識に照らし、抗ヒトIL-4抗体であるREGN668が奏功することは当業者が予測できたことである」
と主張した。
しかし、裁判所は、
「アトピー性皮膚炎の免疫経路の複雑さも考慮すると、炎症部位や病期によってTh1/Th2バランスが変化し、このバランスのみでアレルギー疾患を理解することは困難であったことが本件特許の優先日当時の技術常識であり、それ以前に、IL-4及びこれを産生するTh2細胞を含む、特定の細胞とサイトカインがアトピー性皮膚炎で果たす役割についての当業者の理解は、標的療法の開発の機会を生み出す(特定の細胞とサイトカインを標的に、候補化合物を探索し得る。)にとどまり、特定の細胞とサイトカインのうちのいずれかを標的とすることによって、アトピー性皮膚炎の治療が可能になるような化合物(抗体等)の存在を解明するには至っていなかったといえる。
そうすると、たとえ上記優先日前に、アトピー性皮膚炎の治療が可能になるような化合物(抗体等)の標的となり得る抗原である特定の細胞とサイトカイン(Th2/IL-4)が知られていたとしても、他の多くの細胞とサイトカインも作用することが知られている中で、Th2/IL-4の働きを阻害することで、本件患者を含む慢性アトピー性皮膚炎の治療効果を奏するかどうかまで、当業者が認識できたとはいえない。
つまり、当該抗原の作用を阻害するための受容体に対する抗体(抗IL-4R抗体)が公知であったとしても、当該作用の阻害により、アトピー性皮膚炎の治療効果が可能となるとの治験までが公知になっていたわけではないから、当該抗体(抗IL-4R抗体)を実際に治験に使用して、アトピー性皮膚炎に対する効果を確認してみなければ、アトピー性皮膚炎への治療効果があるかは予測できなかったといえる。
・・・
甲1における試験段階は第Ⅱ相試験であり、甲21によれば、第Ⅰ相試験(フェーズ1)からの移行の成功率は63.2%(n=3,582)であり、第Ⅱ相試験(フェーズ2)から第Ⅲ相試験(フェーズ3)への移行の成功率は更に低く、30.7%(n=3,862。アレルギー疾患の場合には33%)にすぎないことが認められる。しかも、甲1に記載された情報は臨床試験のプロトコル(試験実施計画書)にすぎず、実際の試験結果については記載されていない。そうすると、甲1に記載された治験薬が、試験結果をみるまでもなく当然に治療上有効であると当業者が理解するともいえない。」
と判断した。
これに対し、原告は、
「本件訂正発明1と引用発明の相違点に係る「治療上有効な量」については、薬効を確認するための臨床試験(甲1)において試験される治験薬の量は治療上有効な量であることは当然であり、ヒト抗体のアトピー性皮膚炎に対する具体的な用量は引用発明の際公知であった甲3に記載されている」
などと主張した。
しかし、裁判所は、
「上記主張を踏まえたとしても、Th2/IL-4の働きを阻害すること自体が確たるものではなかったのであるから、本件患者を含む慢性アトピー性皮膚炎の治療効果を奏するかについて当業者が認識できたとはいえないとの前記判断は左右されない。しかも、甲3に記載された「1回あたりの用量は通常約0.01~約20mg/kg体重」の記載は、IL-4Rが関与している種々の病態の処置及び病気に用いる際の用量の目安であって、本件訂正発明の対象患者である本件患者に対する用量は何ら示されていない。」
と述べ、原告の上記主張は採用することができないと判断した。
また、原告は、
「甲1には治験薬としてREGN668が示されているところ、治験薬と医薬組成物は相違しないし、第Ⅰ相試験を経てさらなる臨床試験である第Ⅱ相試験に供試された治験薬組成物に有用性が期待できることは当然であり、さらに、甲2にはこれを「医薬(medications)」であるとする記載がある」
と主張した。
しかし、こちらの主張についても、裁判所は、
「甲1にはREGN668が本件患者に対し治療効果を奏し医薬用途に使用できることについて何ら記載がない。また、甲2においても、「2つの医薬について、ADに対する臨床試験が、IL-4受容体を標的にして現在行われている(Aeroderm及びREGN-668)」(訳)との記載があるだけであり、これは「REGN-668」を含む2つの医薬についてアトピー性皮膚炎に対する臨床試験が行われているという文脈で「医薬」の用語が使用されていると理解されるから、医薬用途に使用できるものとして記載されているとはいえない。」
と述べ、「医薬」の構成に関する原告の前記主張も採用することができないと判断した。
裁判所は、取消事由1に関する原告の主張はいずれも採用できず、本件訂正発明1が、引用発明及び甲1等の記載に基づいて、当業者が容易に発明をすることができたものでないとした本件審決の判断に誤りは認められないと判断した。
(2)取消事由2(サポート要件違反)について
原告は、
「本件明細書に開示された薬理試験結果はmAb1に関するもののみであるところ、本件訂正発明はmAb1とは結合親和性や薬物動態が異なる抗体等を含むものであり、これが臨床で治療に使用可能であるとは当業者は認識しない、その結果、本件特許の権利範囲は本件明細書の開示と比して著しく過大となっている」
から、サポート要件の適合性に関する本件審決は誤りであると主張した。
しかし、裁判所は、以下のとおり、原告主張のサポート要件違反は認められないと判断した。
「本件明細書の記載及び技術常識を総合すると、本件明細書には、①mAb1は、抗IL-4Rアンタゴニスト抗体であって、IL-4Rに結合し、IL-4のシグナルを遮断する作用を有するものであること、②mAb1が投与された本件患者では、アトピー性皮膚炎における臨床症状が改善したこと、③mAb1が投与された本件患者では、アトピー性皮膚炎のバイオマーカーであり、IL-4によって産生・分泌が誘導されることが知られているTARC及びIgEのレベルが低下したことが開示されていることから、これに接した当業者は、本件患者にmAb1を投与した際のアトピー性皮膚炎の治療効果は、mAb1のIL-4Rに結合しIL-4を遮断する作用、すなわち、アンタゴニストとしての作用により発揮されるものと理解するものといえる。
そうすると、IL-4Rに結合しIL-4を遮断する作用を有する抗IL-4Rアンタゴニスト抗体(本件抗体等)であれば、mAb1に限らず、本件患者に対して治療効果を有するであろうことを合理的に認識でき、前記(2)に記載した本件訂正発明の課題を解決できるとの認識が得られるものと認められる。
ところで、本件明細書に開示された薬理試験結果はmAb1に関するもののみであることは、原告の指摘するとおりである。
しかし、サポート要件の適合性につき、「特許請求の範囲に記載された発明が、発明の詳細な説明に記載された発明で、発明の詳細な説明の記載により当業者が当該発明の課題を解決できると認識できる範囲のものであるか否か」等を判断するに当たって、どの範囲の実施例等の裏付けをもって十分とするかについては、当該課題解決の認識がいかなるロジックによって導かれるかという点を踏まえて検討されるべきであり、特許の権利範囲に比して実施例が少なすぎるといった単純な議論が妥当するものではない。
これを本件についてみるに、本件においては、・・・(略)・・・が開示されていることから演繹的に導かれる推論として、本件患者にmAb1を投与した際のアトピー性皮膚炎の治療効果は、mAb1のIL-4Rに結合しIL-4を遮断する作用、すなわち、アンタゴニストとしての作用により発揮されるものと理解されるものであって、課題を解決できると認識できる範囲が幅広い実施例から帰納的に導かれる場合とは異なる。上記作用機序は、本件抗体の一つであるmAb1がIL-4Rに結合し、IL-4のシグナルを遮断する作用を有するものであり、mAb1が投与された本件患者では、アトピー性皮膚炎における臨床症状が改善し、アトピー性皮膚炎のバイオマーカーも低下したのであるから、mAb1以外の抗IL-4Rアンタゴニスト抗体である本件抗体等(mAb1以外の32種)も同様の作用効果を有すると当業者が理解できることは明らかである。
本件明細書に開示された薬理試験結果はmAb1に関するもののみであるとの原告の指摘は、上記認定判断を左右するものではない。
また、原告は、サポート要件違反の根拠として、本件抗体等には、結合親和性、血中半減期、保存安定性等が全く異なるものが含まれている点を挙げる。しかし、アトピー性皮膚炎に対する治療に必要な効果が得られる本件抗体等のスクリーニングが必要となることはあっても(この点は実施可能要件の問題として後述する。)、結合親和性、血中半減期、保存安定性等の違いが、上記作用機序を否定するようなものであると認めるに足りる証拠はない。
したがって、本件抗体等の中には結合親和性等の点で違いが存在するとしても、上記・・・で説示したところに照らして、サポート要件違反を導くものとはいえない。 」
(3)取消事由3(実施可能要件違反)について
原告は、
「①本件特許の特許請求の範囲に記載されている抗体等には、結合親和性が弱いため治療に使用できないものがあり、臨床で治療に使用可能なものを選別しなければならず、また、②治療上の有効量についても、都度臨床試験で確認する必要があり、いずれについても過度の試行錯誤を要するから、本件訂正発明1について実施可能要件違反である」
と主張した。
しかし、裁判所は、以下のとおり、本件訂正発明1について実施可能要件違反をいう原告の主張は採用することができないと判断した。
「原告の主張①についてみると、・・・当業者であれば、本件明細書の発明の詳細な説明の記載及び出願時の技術常識に基づいて、IL-4Rに結合しIL-4を遮断する作用を有する抗IL-4Rアンタゴニスト抗体、すなわち本件訂正発明1における抗体を、公知の方法及びスクリーニングすることにより、過度の試行錯誤を要することなく製造することができ、それを、本件患者に対して投与した場合に治療効果を有することを合理的に理解できるものと認められる。
したがって、本件明細書の発明の詳細な説明は、当業者において、その記載及び出願時の技術常識に基づいて、過度の試行錯誤を要することなく、本件訂正発明1を実施できる程度に明確かつ十分に記載されているといえる。
次に、原告の主張②(治療上の有効量を都度確認する必要があるとの点)を検討するに、本件明細書には、mAb1の具体的用量300mg(実施例10)が開示されており(【0353】)、段落【0019】等にも用量の目安の記載があるから、mAb1以外の抗体についても、アンタゴニスト活性の程度に応じて治療上有効量を設定することが当業者にとって過度の試行錯誤を要するとまで認めることはできない。 」
4.コメント
(1)本件特許が保護する製品
本件特許(第6353838号)の特許権者であるリジェネロン及びサノフィ(被告ら)は、デュピルマブ(dupilumab)を有効成分とするデュピクセント®皮下注に関して以下の本件特許に係る特許権存続期間延長登録出願(審査中)を行っていることから、本件特許は、デュピクセント®皮下注のアトピー性皮膚炎に関する効能効果を保護するものであると推定される。本件明細書の実施例に記載されている抗IL-4R抗体「mAb1」は、REGN668(開発コード名)であり、デュピルマブ(一般名)と同一の物質である。
- 特願2023-700401・・・デュピクセント皮下注200mgシリンジ/既存治療で効果不十分なアトピー性皮膚炎
- 特願2023-700402・・・デュピクセント皮下注300mgシリンジ/既存治療で効果不十分なアトピー性皮膚炎 (生後6カ月以上の小児にはデュピルマブ(遺伝子組換え)として体重に応じて以下を皮下投与する。 5kg以上15kg未満:1回200mgを4週間隔 15kg以上30kg未満:1回300mgを4週間隔 30kg以上60kg未満:初回に400mg、その後は1回200mgを2週間隔 60kg以上:初回に600mg、その後は1回300mgを2週間隔)
- 特願2023-700403・・・デュピクセント皮下注300mgペン/既存治療で効果不十分なアトピー性皮膚炎 (生後6カ月以上の小児にはデュピルマブ(遺伝子組換え)として体重に応じて以下を皮下投与する。 5kg以上15kg未満:1回200mgを4週間隔 15kg以上30kg未満:1回300mgを4週間隔 30kg以上60kg未満:初回に400mg、その後は1回200mgを2週間隔 60kg以上:初回に600mg、その後は1回300mgを2週間隔)
デュピルマブ(dupilumab)は、IL-4受容体複合体及び IL-13受容体複合体に共通の IL-4受容体(IL-4R)αサブユニットに特異的に結合することにより、IL-4、IL-13のシグナル伝達を阻害する遺伝子組換えヒト型モノクローナル抗体である。
日本では、2018年1月19日に「既存治療で効果不十分なアトピー性皮膚炎」の適応でデュピクセント®皮下注300mgシリンジの製造販売が初承認となり、その後、効能又は効果追加、用法及び用量変更追加として、気管支喘息(既存治療によっても喘息症状をコントロールできない、重症又は難治の患者に限る)(2019年3月26日)、鼻茸を伴う慢性副鼻腔炎(既存治療で効果不十分な患者に限る)(2020年3月25日)、既存治療で効果不十分な結節性痒疹(2023年6月26日)、既存治療で効果不十分な特発性の慢性蕁麻疹(2024年2月9日)、生後 6 カ月以上の小児アトピー性皮膚炎の用法・用量(2023年9月25日)が承認された。サノフィ株式会社が製造販売元であり、リジェネロン・ジャパン株式会社が販売提携している。
再審査期間は以下のように設定されている。
- アトピー性皮膚炎:8年(2018年1月19日~2026年1月18日)
- アトピー性皮膚炎の小児用法・用量:4年(2023年9月25日~2027年9月24日)
- 気管支喘息:(2019年3月26日~2026年1月18日)
- 鼻茸を伴う慢性副鼻腔炎:(2020年3月25日~2026年1月18日)
- 結節性痒疹:4年(2023年6月26日~2027年6月25日)
- 特発性の慢性蕁麻疹:4年(2024年2月9日~2028年2月8日)
2023年のデュピクセント®(米国での販売名はDupixent®)のグローバル売上高は、2022年比で33%増の116億ドルに達している(2024.02.02 Regeneron press release: Regeneron Reports Fourth Quarter and Full Year 2023 Financial and Operating Results)。
なんと、デュピクセント®は2023年世界売上トップ6の医薬品だったようです。
(2)科研製薬が無効請求した理由
本件訂正発明1の記載からわかるように、本件特許に係る特許権の効力は、デュピルマブのみならず、抗IL-4R抗体を有効成分とするアトピー性皮膚炎治療薬にも及び得る。
科研製薬は、スイスのNumab Therapeutics AG (以下「ニューマブ社」)と、アトピー性皮膚炎を対象として、IL-4RαとIL-31に結合する二重特異性抗体「NM26」(開発コード NM26-2198)について、日本を含むアジアでのライセンス・共同開発契約を締結し(2021年1月12日)、開発を進めていた。
科研製薬が本件特許に対して無効審判を請求した背景には、アトピー性皮膚炎治療薬として開発中のNM26-2198に関して日本で将来承認に至ったとき、その製品の製造販売に対してリジェネロン及びサノフィ(被告ら)から本件特許に基づいて権利行使されるリスクを問題視しての対応だったと想像される。
しかし、科研製薬による本件特許に対する無効請求は、審決の取消しを求めた本件訴訟でも裁判所に認められることはなかった。
本件特許の20年の存続期間満了日は2033年9月4日(前述のとおり、存続期間延長登録出願手続きは審査中)であり、まだずっと先の将来までその特許障壁は存在し続けるわけだが、科研製薬の2025年3月期第1四半期決算 決算短信参考資料(2024.08.07)によると、NM26の開発ステージもまだフェーズ1とあり、承認までの道程はまだ不透明な状況ともいえる。
本件訴訟の判決が言い渡される約2か月前の2024年5月29日、科研製薬は、NM26について、Janssen Pharmaceutical Companies of Johnson & Johnson(以下「J&J社」)の関連会社との間で知的財産譲渡及び販売提携オプション契約を2024年5月28日付で締結したと発表した(2024.05.29 科研製薬 press release: 「NM26」の知的財産譲渡及び販売提携オプション契約の締結について)。
この発表によると、科研製薬は、本契約締結に伴い、ニューマブ社との共同開発契約において得た全ての知的財産をJ&J社に譲渡すると共に、ニューマブ社と2021年1月12日に締結したライセンス・共同開発契約を解約し、J&J社から契約一時金2,000万米ドルを2025年3月期中に取得する予定である。また、日本及びアジア(韓国、中国(香港含む)、台湾、シンガポール)での開発の進捗および売上の目標達成に応じたマイルストーン収入の総額として最大で1億3,850万米ドル、ならびにアジアでの売上に応じたロイヤリティ収入をJ&J社より受け取る権利を有し、加えて、J&J社が日本で承認取得する全ての適応症について、販売提携契約を交渉するオプション権を有するとのことである。
一方、ニューマブ社とのライセンス・共同開発契約で定めた権利は、ライセンス・共同開発契約の解約後も存続し、科研製薬は、ニューマブ社より契約一時金として6,600万米ドルを2025年3月期中に取得する予定であり、また、J&J社による開発の進捗に応じたマイルストーン収入の総額として最大で1億1,390万米ドルをニューマブ社より受け取る権利を有しているとのことである。
上記契約締結と同日、J&J社は、アトピー性皮膚炎におけるNM26の全世界における権利を取得するため、ニューマブ社を買収する契約を締結したと発表した(2024.05.28 J&J press release: Johnson & Johnson to Obtain Rights to a Clinical-Stage Bispecific Antibody to Address Distinct Patient Needs in Atopic Dermatitis)。
今後、科研製薬又は今後のNM26の開発を主導すると思われるJ&J社は、本件特許に対してどのように向き合っていくのでしょうか。
(3)ClinicalTrials.govについて
進歩性欠如の無効理由として科研製薬(原告)が挙げた引用文献(甲1)は、臨床試験情報データベースClinicalTrials.govに登録されたデュピルマブの臨床試験(フェーズ2)のプロトコル(試験実施計画書)であった。
もちろん、その登録手続きをしたのは、開発を進めていたリジェネロン及びサノフィ(被告ら)であり、その臨床試験の結果は、本件特許の明細書に実施例として記載された。
ClinicalTrials.govとは、2000年に開設された臨床試験に関する情報を提供する世界最大級のオンラインデータベースであり、米国国立公衆衛生研究所 (NIH) 、米国医薬食品局 (FDA)、米国国立医学図書館 (NLM) が共同で運営している。FDAへの医薬品申請に関連する臨床試験を実施する者はこのデータベースに臨床試験を事前登録し結果を公開することが義務付けられており、これにより、臨床試験の透明性を高め、被験者への倫理的配慮を促進する役割を果たすとともに、臨床試験情報を集積するレジストリとして、患者や医療従事者が進行中の臨床試験に関する情報にアクセスしやすくすることにも役立っている。
出典: https://www.clinicaltrials.gov/about-site/about-ctg
ヘルシンキ宣言(2013改正)では、ヒトを対象とする全ての研究は、最初の被験者を募集する前に公開データベースに登録し、結果は肯定的・否定的に関わらず公表する義務があることを定めている(35条及び36条)。
また、国際製薬団体連合会(IFPMA)、米国研究製薬工業協会(PhRMA)、欧州製薬団体連合会(EFPIA)及び日本製薬工業協会(JPMA)は、「Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databeses(臨床試験登録簿及びデータベースを介した臨床試験情報の開示に関する共同指針)」に則り、自社がスポンサーとなる医薬品の臨床試験情報を公開するとしている。
このように、臨床試験を実施する者には、公衆衛生上の利益として臨床試験の透明性を約束するため、臨床試験を開始する前に、ClinicalTrials.govを代表とする公開データベースにその臨床試験に関する一定の情報を、決められた時期までに、登録し公開することが求められている。この登録・公開は、臨床試験の結果が得られてから初めて行うものではなく、試験を開始する時点から求められている。
欧州連合(EU)での臨床試験については、臨床試験情報システム(the Clinical Trials Information System(CTIS))が稼働しましたね。
(4)臨床試験と出願のタイミング
臨床試験開始時からその情報公開が求められているところ、特許権者であるリジェネロン及びサノフィは、臨床試験結果に基づく医薬用途発明に係る本件特許の出願をどのようなタイミングで踏み切ったのか考えてみたい。
みなさんなら、この臨床試験を開始するに際して、どのタイミングで出願しますか?
本件明細書には、中等度から重度のアトピー性皮膚炎を有する患者に「mAb1」(REGN668(開発コード名)、デュピルマブ(一般名)と同一物質)を投与した臨床試験結果が実施例6から実施例11として記載されている。それら各実施例は、試験設計等の記載から、ClinicalTrials.govに登録された以下の臨床試験に該当すると推測できる。なお、実施例10が、引用文献として挙げられた甲1の臨床試験プロトコル(NCT01548404)の結果も含むものである。
- 実施例6:NCT01259323 “Sequential Ascending Dose Study to Assess the Safety and Tolerability of REGN668 (SAR231893) in Patients With Atopic Dermatitis” (Phase 1b)
- 実施例7:NCT01385657 “Safety and Tolerability of Dupilumab in Participants With Moderate to Severe Atopic Dermatitis” (Phase 1b)
- 実施例8:実施例6及び実施例7を合わせた分析
- 実施例9:NCT01859988 “Study of Dupilumab Administered to Adult Patients With Moderate-to-Severe Atopic Dermatitis” (Phase 2)
- 実施例10:NCT01548404 “Study of Dupilumab in Adult Patients With Extrinsic Moderate-to-Severe Atopic Dermatitis” (Phase 2)
- 実施例11:NCT01639040 “Study to Assess the Safety of Dupilumab (REGN668/SAR231893) Administered Concomitantly With Topical Corticosteroids (TCS) in Patients With Moderate-to-severe Atopic Dermatitis (AD)” (Phase 2)
2013年3月2日に、リジェネロン及びサノフィは、アトピー性皮膚炎におけるデュピルマブのポジティブなPoC(Proof of concept)データを得たことをプレスリリースにて発表した(2013.03.02 Regeneron press release: Sanofi and Regeneron Report Positive Proof-of-Concept Data for Dupilumab, an IL-4R alpha Antibody, in Atopic Dermatitis)。これは、本件特許の明細書に実施例8として記載されたNCT01259323及びNCT01385657を合わせた分析結果についての発表である。
本件特許に係る出願日は、2013年9月4日であるが、2012年9月7日にした米国仮出願を最先として計9つの出願に基づく優先権を主張している。
本件特許出願に係る優先権の基礎となる各出願の明細書の実施例に盛り込まれたこれらアトピー性皮膚炎に対するデュピルマブの臨床試験登情報の対応関係とともに、臨床での有効性を初めて公表したプレスリリースとの時間軸を以下の図にまとめた。
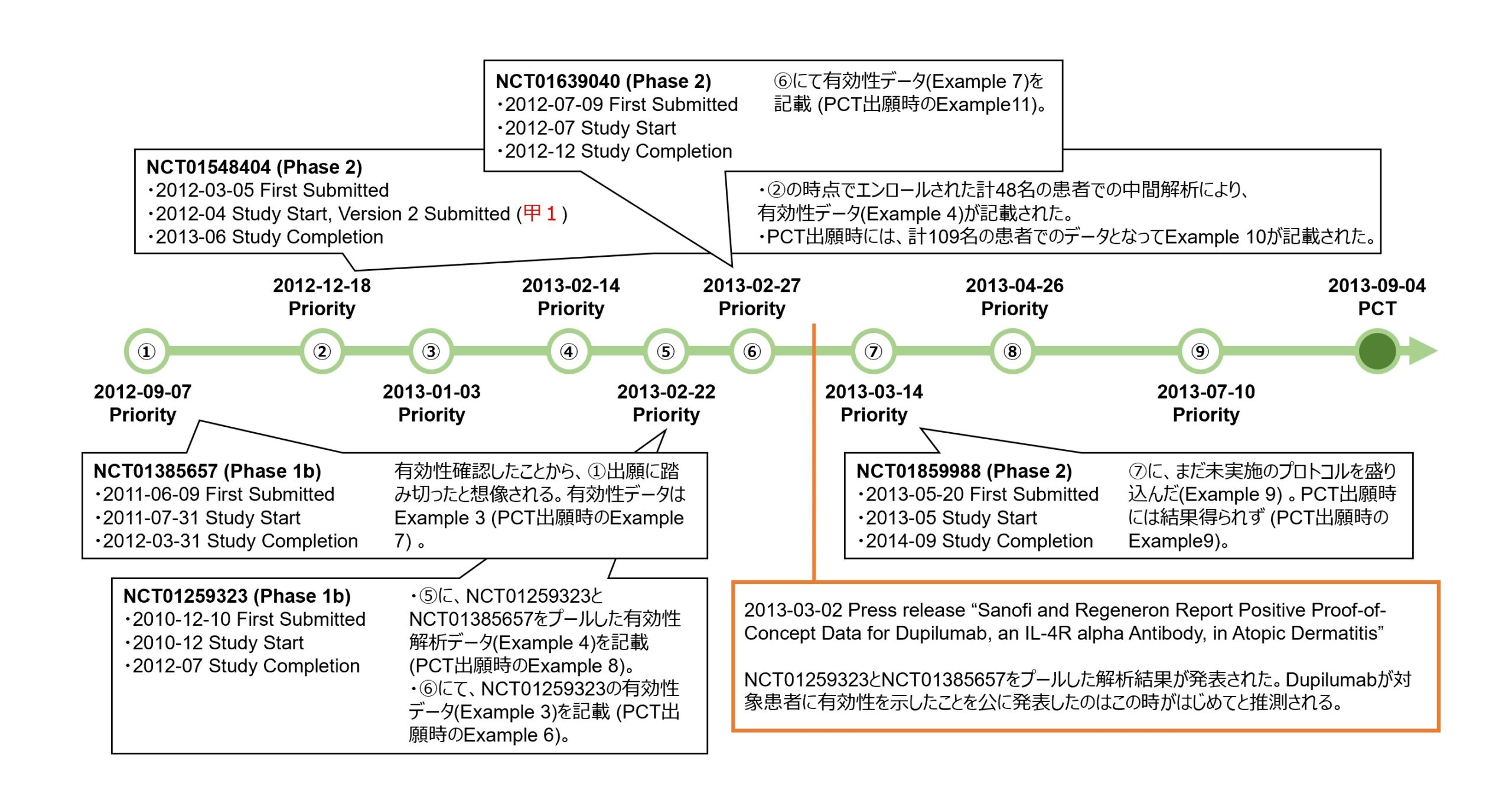
臨床試験の登録と出願との時間軸を眺めると、アトピー性皮膚炎患者を対象とする臨床試験では、2010年12月10日に、NCT01259323(Phase 1b)の試験開始を登録する最初の情報が提出された。
しかし、それよりも、2011年6月9日に提出されたNCT01385657(Pase 1b)の臨床試験が進捗し、中等度から重度のアトピー性皮膚炎患者に対してデュピルマブの皮下投与が有効性を示したデータが取得されたこと(2012年3月にStudy Completion)を契機として、本件特許に係る出願の優先権の基礎となる最初の米国仮出願(図中①)が2012年9月7日に行われたようだ。この出願①の明細書には、同臨床試験のプロトコルや有効性を示すデータが実施例3として記載された(後のPCT出願では実施例7)。
さらに、2012年12月18日、NCT01548404(Phase 2)の中間解析と思われる計48名の患者における有効性データが盛り込まれた米国仮出願(図中②)が行われた。この臨床試験は2013年6月に完了し、計109名の患者でのデータとなってPCT出願時に実施例10として記載された。2012年4月にClinicaltrials.govに提出されたこの臨床試験プロトコルの登録情報の公開が、本件訴訟で進歩性欠如の有無の判断において問題となった引用文献(甲1)である。
その後、臨床試験の進捗に応じて、優先権の基礎となるように米国仮出願(図中③から⑧)及びフランス出願(⑨)が次々と行われ、臨床試験のプロトコルや結果に関する実施例の記載が充実されていった。
デュピルマブがアトピー性皮膚炎患者に対して有効性を示したことが公に初めて発表されたのは、2013年3月2日のプレスリリースのタイミングだったと思われる(図中、2013-03-02 Press release)。その発表内容であるNCT01259323(Phase 1b)とNCT01385657(Phase 1b)をプールした解析から得られた有効性データ(後のPCT出願では実施例8)は、そのプレスリリースの約1週間前(2月22日)にされた出願⑤、そしてNCT01259323(Phase 1b)単独のデータ(後のPCT出願では実施例6)及び局所コルチコステロイドとの併用効果を示したNCT01639040(Phase 2)のデータ(後のPCT出願では実施例11)は2月27日にした出願⑥に盛り込まれるなど、発表直前に実施例の記載が充実されていったことがわかる。
上記のとおり、本件特許に係る出願の優先権の基礎となった各出願のタイミングから言えることは、この出願戦略は、臨床試験結果が得られてからそのデータを記載して出願する記載要件重視型であるといえる。これが、もし、新規性・進歩性重視の出願戦略を選んでいたとしたら、NCT01259323(実施例6に該当)の臨床試験開始時の最初の登録手続き(2010年12月10日)の前というタイミングで仮出願することになっていただろうが、そうなればタイミング的にその臨床試験結果であるPoCデータを得ることができないままPCT出願をせざるを得ないことになっていただろうし、結果として、実施可能要件及びサポート要件を欠いたPCT出願となっていただろう。
(5)進歩性の判断について
臨床試験プロトコルの公開とその臨床試験結果に基づく本件特許出願との関係を踏まえ、臨床試験プロトコルの公開情報(甲1)が引用文献となり進歩性が問われた本件特許の裁判所の判断について考えてみたい。
裁判所は、甲1の「治験薬」と本件訂正発明1の「医薬」との間には相違点があると認定したうえで、甲1における試験段階は第Ⅱ相試験(フェーズ2)であり、その成功率の低さ、さらには甲1に記載された情報は臨床試験のプロトコルにすぎないことからすると、甲1に記載された治験薬が、試験結果を見るまでもなく当然に治療上有効であると当業者が理解するとはいえないと判断した。
審決及び判決は、以下の甲21(審判乙1)により、第II相試験(フェーズ2)の成功率は低く、アレルギー疾患の場合には33%にすぎないと認定している。
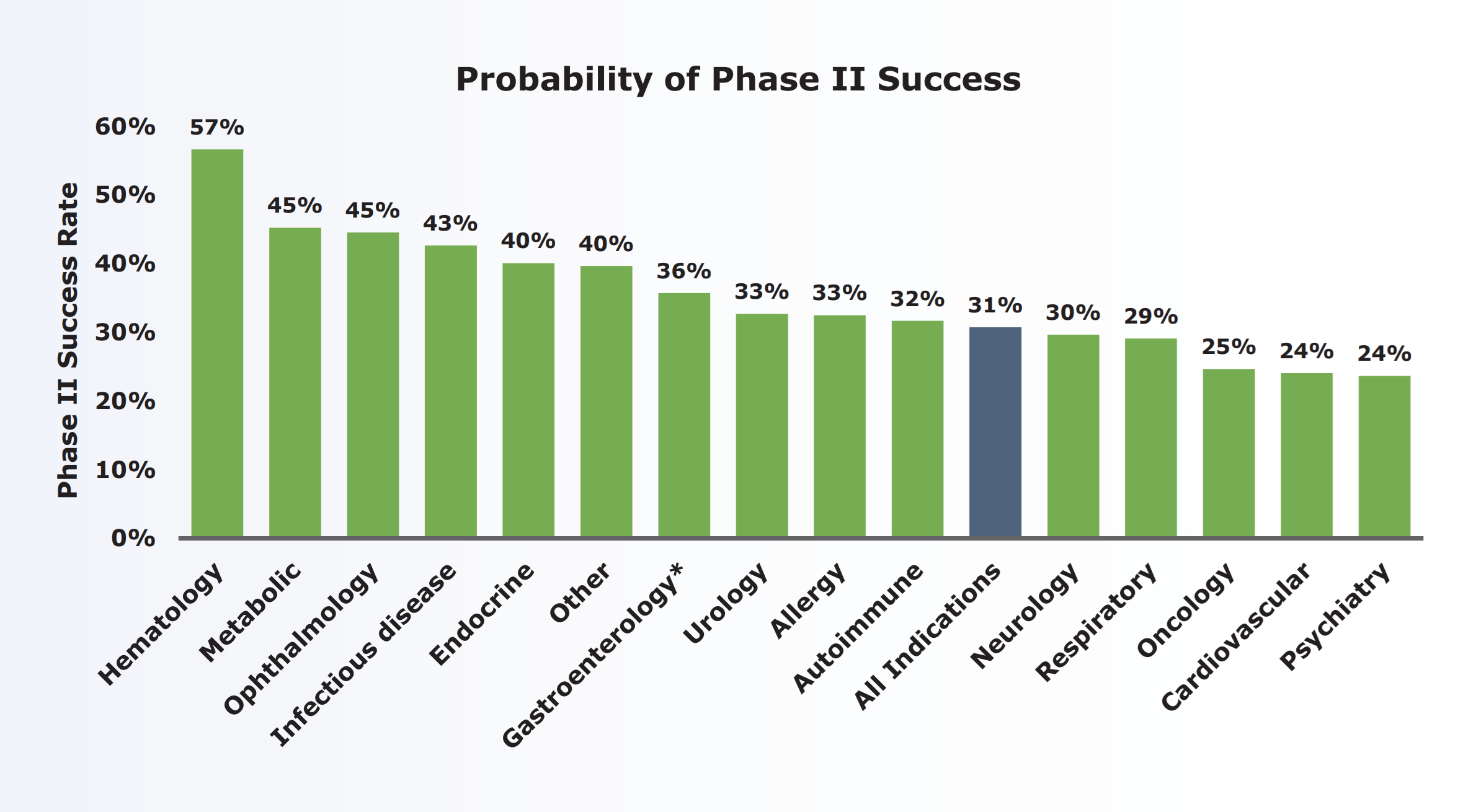
出典: 無効2021-800003 審決文 p25 乙1ウ10頁図3
本件審決及び判決の認定によると、医薬用途発明の新規性及び進歩性の判断において、一般的には、当該発明と臨床試験実施中である旨(例えばプロトコル)が記載された引用発明との間には、「医薬」と「治験薬」という相違点があることになる。
そして、臨床試験(治験)の成功率に加えて、「実際に治験に使用して、アトピー性皮膚炎に対する効果を確認してみなければ、アトピー性皮膚炎への治療効果があるかは予測できなかった」、「治験薬が、試験結果をみるまでもなく当然に治療上有効であると当業者が理解するともいえない」、「Th2/IL-4の働きを阻害すること自体が確たるものではなかった」といった言葉からも、裁判所は、進歩性欠如の主張を認めるには、引用発明に効果の実証的記載又は技術常識を総合して当業者が効果を予測できる程度の確たる合理的理由が必要であることを求めているといえる。
裁判所のこの求めに関しては、特に、既存の医薬品とは異なり新たな作用機序であって従来の治療体系を大幅に変えるような独創的医薬品を意味する「ファースト・イン・クラス」を狙う開発の場合には、臨床試験結果を見るまでもなく期待する治療効果を示すだろうと当然に予測ができること(容易想到性)を示すことは、ますます難しいのではないかと思われる。
デュピルマブは、2017年3月28日、中等度から重度の成人アトピー性皮膚炎患者に対して有効性を示す生物学的製剤(抗IL-4R抗体)として初めてFDAから承認を取得したことから、ファースト・イン・クラス・ドラッグとされている(FDA CDER report: Advancing Health through Innovation: 2017 New Drug Therapy Approvals)。
ここで、注意が必要なことは、臨床試験の成功率の低さから、「治療効果を示すかどうか臨床試験結果を見ずに予測することなんて誰もできるはずがない・・・」からといって、臨床試験結果に基づく医薬用途発明について、一律に「臨床試験プロトコルが情報開示されてから出願しても進歩性は大丈夫!」というわけではないことである。臨床試験のプロトコルや試験実施中である旨を報じた文献が新規性又は進歩性を否定することになるかどうか争われた事例として、以下の判決がある。大半は、新規性又は進歩性が否定された判決である。
- 2007.03.01 「ブリストルマイヤーズスクイブ v. 日本ケミカルリサーチ」 知財高裁平成17年(行ケ)10818(臨床試験プロトコルの記載により新規性を否定)
- 2013.10.16 「沢井製薬 v. 第一三共」 知財高裁平成24年(行ケ)10419(医薬発明が開示されているといえるためには薬理試験が医薬用途を合理的に推論できる試験であれば足りるとして進歩性を否定)
- 2017.02.28 「ザ・ヘンリー・エム・ジャクソン・ファンデイション v. 特許庁長官」 知財高裁平成28年(行ケ)10107(臨床効果が記載されていないから引用発明の認定を否定)
- 2021.06.29 「EAファーマ v. 沢井製薬・大原薬品工業」 知財高裁令和2年(行ケ)10094(臨床試験計画(プロトコル)の記載と技術常識から進歩性を否定)
本願発明の属する技術分野における出願時の技術水準を的確に把握した上で、引用発明に基づいて当業者が請求項に係る発明に容易に想到できたどうかが、進歩性判断の基本的な考え方であることには変わりないが、臨床試験プロトコルの公開情報である引用文献とその臨床試験結果に基づく医薬用途発明に係る特許性との関係において、本件判決は、引用文献に実証的記載がなかったことにより進歩性が認められた事案として示唆のあるものであった。
しかし、その判断は、臨床試験プロコトル公開情報の引用発明としての位置付けを一般化できるようなものではなく、抗IL-4R抗体と対象患者における出願当時の技術常識を背景とした本件特有の事情に大きく依存していたように思われる。
引用文献は臨床試験プロトコルではないが、医薬用途発明の進歩性を巡って、効果等の実証的データの記載がない引用文献が問題となった判決も多数あり参考になる。
- 2007.07.12 「エンシステックス v. バイエルクロップサイエンス」 知財高裁平成18年(行ケ)10482(引用文献に具体的な生物試験の結果が示されていなかったが、適用してみようとすることは何ら困難な事柄ではないとして進歩性を否定)
- 2008.07.30 「ファルマシア・アンド・アップジョン v. 特許庁長官」 知財高裁平成19年(行ケ)10377(著者が根拠のない単なる希望や空想ではなく専門家として見解を記載していると考えるのが自然であり、試験結果の記載があるかどうかにより左右されるものではないとして進歩性を否定)
- 2008.11.26 「バイエル v. 大洋薬品」 東京地裁平成19年(ワ)26761(文献には製造方法が記載されていないが刊行物に記載された発明であると認定され新規性を否定)
- 2012.04.11 「沢井製薬 v. 武田薬品」 知財高裁平成23年(行ケ)10148(引用例の図には本件発明の構成が記載されており、その作用効果又は作用効果に関わる構成も記載されているに等しいというべきであって、刊行物に記載された発明であるというほかないと判断)
- 2014.08.07 「セルジーン v. 特許庁長官」 知財高裁平成25年(行ケ)10170(一行記載の疾患にも合理的に有効であると理解できると判断)
- 2015.08.20 「サントリー v. 特許庁長官」 知財高裁平成26年(行ケ)10182(用途の一行記載(一例記載)を認定せず、実施例で示された疾患の治療を認定)
- 2016.03.08 「キュアバック v. 特許庁長官」 知財高裁平成27年(行ケ)10043(技術的思想を実施し得る程度に技術的思想の内容が開示されていることが必要であり、かつ、それで足りる)
- 2021.03.25 「沢井製薬 v. 東レ」 知財高裁令和2年(行ケ)10041(技術的な裏付けの乏しい一つの仮説にすぎないとして動機付けを否定)
- 2023.01.12 「東和薬品・共和薬品工業・日医工 v. 協和キリン」 令和3年(行ケ)10155, 10157・・・パーキンソン病治療剤ノウリアストの医薬用途発明の特許性 引用文献記載は「実証的でない」と判断(引用文献は「これを裏付ける試験結果等に基づいて実証的な記載であるということはできない」として新規性・進歩性を肯定。)(『医薬系特許的判例ブログ年報 2023』 Fubuki著 2024年5月発行, p28-52)
(6)欧米ファミリーの状況
本件特許(第6353838号)は、9件のパリ条約による優先権を主張して2013年9月4日に出願されたPCT国際出願(出願番号PCT/US2013/057898; 公開番号WO2014/039461)から日本に移行した特許出願に係るものである。
特許として登録されれば、出願日から20年の存続期間満了日は2033年9月4日となる。
日本では、臨床試験プロトコルの登録情報の公開により進歩性欠如が争われたが、結果的には、臨床試験結果が得られてからそのデータを記載して出願する記載要件重視型出願戦略を選択したことが、功を奏した。
では、同特許ファミリーである欧米での出願は特許性に関してどのように判断されたのだろうか。
以下に紹介するが、米国は日本に比べて厳しい特許性判断がされていることがわかる。特許性判断において、米国では、引用文献における効果の実証的記載の有無やその効果の合理的推論の有無よりも、その臨床試験を実施していること自体が新規性において致命的となった。
ア 米国の状況
米国では、本件特許のファミリーである14/017,333(公開番号US2014-0072583A1)が出願された後、登録には至らず放棄され、継続出願15/610,267(公開番号US2017-0333557A1)で権利化を試みるもこれも放棄し、現時点では、継続出願である17/951,987(公開番号US2023-0058395A1)が審査に係属している。米国では特許性について厳しい判断がされている。
14/017,333(公開番号US2014-0072583A1)での審査において、出願人は、抗IL-4R抗体を投与することによるアトピー性皮膚炎の痒みを低減させる方法に関するクレームでの権利化を試みたが、最終的には、記載要件(112条 first paragraph – written description)、非自明性(103条)及びダブルパテンティングに基づく拒絶を受け、放棄した。非自明性を否定された際に挙げられた先行技術は、Martinらの文献 (US7,608,693又はUS8,337,839)等であった。
“Given the combined teachings of the ‘693 patent and Morioka, one of ordinary skill in the art would have had the motivation to treat AD by administration of an anti-IL-4R antibody. The ‘693 patent discloses a specific anti-IL-4R antibody, and teaches that this antibody is useful for treatment of IL-4-mediated disorders, including AD. Further motivation comes from Morioka, which shows that AD can be treated by suppression of IL-4 signaling. Although it is acknowledged that Morioka administered a nucleic acid which encoded a mutant IL-4 molecule which functions as an IL-4 antagonist, rather than administering an anti-IL-4 antibody, it nevertheless shows that suppression of IL-4 signaling is effective for treatment of AD. One of ordinary skill in the art would have had a reasonable expectation that the anti-IL-4R antibody of the ‘693 patent would also inhibit IL-4 signaling, as this antibody, like the IL-4 antagonist of Morioka, specifically binds a human IL-4R. Therefore the skilled artisan would have had the motivation, and a reasonable expectation of success, to select AD as a specific disorder than can be treated by administration of the anti-IL-4R antibody of the ‘693 patent.”
継続出願15/610,267(公開番号US2017-0333557A1)の審査では、CDRを特定した具体的な抗IL-4R抗体を投与することによるアトピー性皮膚炎の痒みを低減させる方法に関するクレームの権利化を試みたが、Martin(US8,337,839B2)、Ong(Editorial update on emerging treatments of atopic dermatitis, 17 Expert Opin. Emerging Drugs 17:2 129-133 (2012)、2012年3月11日公開)、Radin(US10,485,844B2)等の組み合わせに基けば自明であること(103条)から特許性無しとする最終拒絶を受け、出願人は審判を請求した(Appeal 2021-004563)(クレーム1は下記のとおり)。
1. A method for reducing pruritus associated with atopic dermatitis (AD) in a patient, the method comprising:
(a) selecting a patient with moderate-to-severe atopic dermatitis, wherein the patient exhibits a Pruritus Numeric Rating Scale (NRS) score ≥5 and/or a 5-D Pruritus score ≥18; and
(b) administering to the patient one or more doses of a pharmaceutical composition comprising a therapeutically effective amount of an anti-interleukin-4-receptor (IL-4R) antibody or antigen-binding fragment thereof; wherein the antibody or antigen-binding fragment thereof comprises three heavy chain complementarity determining regions (HCDRs) comprising …
しかし、米国特許商標庁(USPTO)の特許審判部(Patent Trial and Appeal Board (以下「PTAB」))は、MedChemExpress(https://www.medchemexpress.com/dupilumab.html, last accessed July 8, 2022)により裏付けられたOng、Radin等によれば新規性が欠如しているという新たな拒絶理由(102条(a))も加えて拒絶決定を支持する判断を下した。
Ongは、抗IL-4R抗体であるREGN-668が、中等度から重度のアトピー性皮膚炎患者の治療のためのフェーズ1/2試験(Randomized, double blind, placebo controlled, sequential ascending, repeated dose; moderate to severe AD.)が実施中であることを開示しており、Radinは、dupilumabとしてクレームのCDRsを開示していた。そして、MedChemExpressは、出願後であるが、dupilumabはREGN-668と同義語であることを開示していた。
“For the forgoing reasons, we find that Ong, which describes the administration of the same antibody required by Appellant’s claim 1 to the same patient population required by Appellant’s claim 1, will inherently reduce pruritus associated with atopic dermatitis in a patient and, therefore, anticipates Appellant’s claim 1.”
米国の審査では、臨床試験を実施していることを開示した文献(その内容はClinicaltrials.govの開示情報由来と想像される)が先行文献として採用され、PTABにより新規性欠如(102(a))を理由に拒絶審決を受けた。
本件特許に係る出願の最先の優先日において、「痒みを低減させる」ことは臨床試験結果として公表されてはいなかったが、アトピー性皮膚炎患者を治療する目的のために臨床試験で使用されていたことが公表されていたという事実は、(結果的には効果があったわけだから)潜在的(inherently)には痒みを減少させている使用が知られたことになり、米国においては、新規性を欠如させる理由として十分だったのかもしれない。
効果があるかどうか結果は得られていなかったとしても、結果的には効果があったわけだしな・・・
本事件のように、臨床試験として実施しているという事実が既に公衆に知られ、その効果確認が待たれる臨床試験により用いられている方法についての公知文献がある場合、いかなる要件の下で同方法発明の特許を認めるべきか(あるいは認めるべきではないのか)という論点は、内在同一の問題にも関係してくるかもしれない(ブログ記事「「内在同一の問題」 -製薬・バイオテクノロジー分野における新たな科学的発見と公衆衛生との間で揺れる特許保護のジレンマ-」参照)。
出願人は、この審決に対して手続きを進めず、新たに再度の継続出願17/951,987(公開番号US2023-0058395A1)を行い、現在、これが審査に係属している。
臨床試験の実施が公然実施(public use)に該当することを理由に新規性欠如(102条(b))となるか争われた事件として、例えば、古くは以下の事件があるが、本事件は、公然実施ではなく臨床試験情報登録公開という先行文献に基づく102条(a)の問題である。
臨床試験と公然実施に関するCAFC判決に関する記事:
- 2008.08.20 「AstraZeneca v. Apotex and Impax」 CAFC Docket No. 2007-1414, -1416, -1458, -1459
- 2006.12.26 「Eli Lilly v. Zenith, Teva, Dr. Reddy’s」 CAFC Docket No.05-1396, -1429, -1430
- 2004.04.23 「SmithKline Beecham v. Apotex」 CAFC Docket No.03-1285
ところで、FDAによるDupixent®(デュピルマブ)の承認(2017年3月28日)に基づいて特許権の存続期間延長が認められたのは、米国特許7,608,693号である。この特許は、承認から14年の上限規定のもと1273日の延長が認められ、満了日は2031年3月28日となっている。また、この特許から分割された特許8,337,839号が、前述の拒絶理由通知で挙げられた引用文献(Martin)に該当する。
仮に、本件特許のファミリーに属する米国出願も特許となっていた場合、20年の存続期間は2033年9月4日まで延びていたはずである。そうであれば、延長が認められた特許7,608,693号の満了日(2031年3月28日)から約2年5か月の追加的な製品保護が期待できたかもしれない(これはあくまで仮定の話である)。一方、新規性の確保を重視して、NCT01259323(フェーズ1b)の臨床試験が始まる直前の2010年12月頃に最初の出願を行っていたとすれば、特許を取得できた可能性はあるが、その場合の満了日は2031年12月頃となり、延長対象特許7,608,693号の満了日(2031年3月)からの保護期間は約9か月と短くなっていただろう。
米国における製品保護期間に与える影響を考えると、臨床試験結果が出てから出願する記載要件重視の出願タイミングは、臨床プロトコルが公開された後の新規性を失うリスクを冒してでも特許取得に成功した場合の製品への利益貢献を重視する選択肢として、ビジネス的には、より有望だったのかもしれない。
イ 欧州の状況
本件特許の欧州ファミリーであるEP2892927Bは、審査では、米国ファミリーの審判でも挙げられたOngらの文献等に基づき進歩性欠如等が指摘されたが、同文献にはクレームされている特定の患者について開示も示唆もされていないことや同文献では”challenging”と述べているように未だ統計学的にも有効性が示されていなかったにもかかわらず本願発明は予期せぬ効果を示せたことなどを出願人が主張(2017年1月6日)したことによって、それら拒絶理由は解消され、登録に至った(クレーム1は下記のとおり)。
同特許については、異議申立てが提出された。
異議申立てにおいて、先行技術文献D1として審査でも挙げられたOngらの文献や、D7として本件明細書中の実施例10に相当する臨床試験登録NCT01548404(Phase 2)の最初のプロトコル開示情報が挙げられなどし、PCT出願時にはSanofiが共同出願人になっていなかったことを理由に優先権主張の利益が得られていない出願である点が主張されたほか、D1(Ong)やD7(NCT01548404)に基づく新規性及び進歩性の欠如等が主張された。
臨床試験実施中と述べた引用文献には特定の患者群に言及されていなかったのが幸いしたか・・・進歩性の判断としてはスレスレだったのでは。
しかし、2019年9月5日に特許権者側から同特許許可を取下げる申し出がなされたことから(理由は不明)、その申し出により、欧州特許庁は同特許を無効とし、異議申立て手続きは終結した。
また、分割出願として下記のクレーム1のEP3889181Bが2024年4月24日に登録された。相補性決定領域(complementary determining regions(CDRs))を特定した具体的な抗IL-4R抗体をクレームしたものとなっており、現時点で異議申立ては提出されていない。
1. An anti-interleukin-4-receptor (IL-4R) antibody for use in a method of treating moderate-to-severe atopic dermatitis (AD) in a patient, the method comprising administering an initial dose of the antibody followed by at least eight secondary doses of the antibody, wherein:
– the initial dose is a loading dose of 300mg or 600mg, followed by the secondary doses which are maintenance
doses of 300mg;
– each secondary dose is administered to the patient 1 to 2 weeks after the immediately preceding dose; and
– the antibody comprises an HCVR having the amino acid sequence of SEQ ID NO: 162 and an LCVR having
the amino acid sequence of SEQ ID NO: 164.
さらに、下記クレーム1のEP4374919Aが分割出願として2024年5月29日に公開されている。親特許に当たるEP3889181Bは異議申立てにより無効と判断されたが、このクレームで抗IL-4R抗体という機能的に表現した用途特許を狙いに行くのかもしれない。
ドイツ、英国、フランスでのデュピルマブの承認に基づいて付与される特許権存続期間延長の補充的保護証明書(Supplementary Protection Certificates: SPCs)について確認すると、対象は欧州特許EP2356151Bである。この特許は2009年10月27日に出願され、2017年9月のEU承認から15年の上限規定のもと、満了日は2032年9月28日である。さらに、小児延長手続きを経て、満了日は2033年3月28日に延長されたか又は延長される予定である。これは、米国で特許期間延長の対象となった米国特許7,608,693と同じファミリーに属する。
また、本件特許ファミリーの欧州特許EP2892927Bは、存続期間が20年で、その満了日は2033年9月4日である。このため、SPC延長によって得られる保護期間終了後も残る欧州特許EP2892927Bの製品保護期間は半年未満となる。仮に、新規性の確保を重視し、NCT01259323(フェーズ1b)の臨床試験開始直前である2010年12月に最初の出願を行っていたとすれば、その場合の満了日は2031年12月頃となり、SPC延長特許が満了する前に保護期間が終了してしまうことから、製品保護としての価値はないことになる。
欧州での製品保護期間に関しても、臨床試験結果が出てから出願する記載要件重視の出願タイミングは、臨床プロトコル公開後に新規性を失うリスクを冒してでも、特許取得に成功した場合、たとえ保護期間が半年ほどであっても、製品への利益貢献を期待できる選択肢だったのかもしれない。
(7)臨床試験結果に基づく特許出願のジレンマ
ここまで、臨床試験プロトコルの情報公開が特許性に与える影響について述べてきた。その影響は、国によって異なり、本事件で見られたように、日米欧で大きな違いが見られた。
医薬用途発明の特許出願を臨床試験結果に基づいて行う場合、新規性や進歩性を確実にするために、臨床試験プロトコルが公開される前(臨床試験の開始直前)に出願しておけば良いのかというと、これまで述べてきたことから、必ずしもそう単純ではないことがわかるだろう。
適応症によって異なるが、治験薬の有効性を確認するための臨床試験は、結果が出るまでに数年かかることもある。もし試験結果が出る前に特許出願を行い、1年以内にその結果を明細書に盛り込むことができなければ、記載要件に問題が生じる可能性もある。
そこで、臨床試験を計画している製薬会社は、臨床試験結果に基づく医薬用途発明の特許出願を行うにあたり、次の二つの選択肢に直面することになる。
- オプション1:臨床試験開始(プロトコル公開)前に出願する(新規性・進歩性重視)。
- オプション2:臨床試験結果が出てから出願する(記載要件重視)。
本件特許に係る出願で、リジェネロンとサノフィは後者の「記載要件重視」のオプション2戦略を採用した。
この戦略により、日本では特許が維持されたが、米国ではPTABにより新規性の欠如が理由で拒絶された。米国では審査中にデータを提出して主張すること(いわゆる「後出しデータ」)が比較的許容されるため、リジェネロンとサノフィは当然ながら事前に「新規性・進歩性重視」のオプション1戦略を採ることも検討していたに違いない。
対照的に、日本では記載要件が厳しく、後出しデータが認められないことが多いため、「記載要件重視」のオプション2戦略が有効だったと考えられる。
このように、国ごとに臨床試験プロトコルの位置づけや後出しデータの許容範囲が異なるため、米国では「新規性・進歩性重視」、日本では「記載要件重視」のように、国に応じた戦略を取るのが望ましいだろう。
あるいは、両方の戦略を併用して、柔軟な権利化の機会を確保する方法も考えられる。ただし、その場合の戦略を練る際には、オプション1の出願公開がオプション2の新規性や進歩性に影響を与える可能性があるため、注意が必要である。
特に日本では、優先権の主張にも注意が必要である。効果実証の記載がない基礎出願では、効果が十分に記載されていないと判断され、優先権が認められない可能性がある。
参考:
- 2023.04.06 「グリーンクロス v. シャイアー」 知財高裁令和4年(行ケ)10010 - 医薬用途発明の優先権の効果が認められるには?/本件特許はイズロン酸-2-スルファターゼ脳室内投与製剤(ムコ多糖症II型治療剤ヒュンタラーゼ®)の障害となるのか? -・・・本件において、優先権の効果が認められるためには、基礎出願に少なくともその医薬用途としての効果が実質的に記載されていると認められる必要があると判示した。優先権の効果が認められるための記載について争われた過去の医薬関連判決を眺めると、医薬用途発明の優先権の効果について真正面から判示した裁判例は少なく、この判決はひとつの基準を示したものといえるかもしれない。(『医薬系特許的判例ブログ年報 2023』 Fubuki著 2024年5月発行, p132-161)
- 2024.03.26 「フマキラー v. アース製薬」 知財高裁令和5年(行ケ)10057 ― 優先権主張の効果、補正・訂正要件、実施可能要件の交差点 ―・・・知財高裁は、「人工乳首事件」東京高裁判決の説示を踏襲しつつ、「後の出願の特許請求の範囲に記載された発明の要旨とする技術的事項が、先の出願の当初明細書等に記載された技術的事項を超える」ものか否かという判断はその関係において「実施可能であるかを判断するものと解される」と一歩踏み込んだ。

国ごとに戦略をカスタマイズしたとしても、すべての国にとってベストな選択肢がない場合は、最終的に米国市場の重要性を考慮して、米国を重視する選択が必要になるかもしれない。
さらに、欧米のように特許延長が原則1つの特許にしか適用されない国では、どの特許を延長すべきか慎重に検討する必要もある。前述の欧米ファミリーの例のように、もし別の特許を延長すると想定している場合、それぞれの特許が製品保護期間にどれほど役に立つのかも、いつ出願するか検討する際には見積もっておかなければならない。
このように、臨床試験結果に基づく医薬用途発明の特許出願のタイミングは、発明の内容や技術常識によってケースバイケースかもしれないが、臨床試験のプロトコル公開から結果が得られるまでの試験計画や進捗に応じて、優先権、新規性、進歩性、記載要件などについての各国の法律と、製品保護期間と範囲等から導き出されるビジネス的観点を総合的に考慮して組み立てる必要がある。
この臨床試験プロトコルの公開が特許性に与える影響に関するジレンマについて詳しく論じた以下の海外レポートがある。
参考:
- 韓国の知的財産権侵害 判例事例集(2021年3月 独立行政法人 日本貿易振興機構ソウル事務所): 2. 抗癌剤併用療法発明の新規性及び進歩性が先行文献としての臨床試験計画書により否定されないとした事例
- The Clinical Trial Dilemma: Update on the EPO Position(Broadcast date: 27 June 2023 — Speakers: Amanda Simons, Tanja Preissner)
- 臨床試験ルーレット:オーストラリアでの医薬品の特許取得は今や運次第(5TH JANUARY 2022 Spruson & Ferguson)
(8)機能的表現抗体の医薬用途発明の記載要件
本件は、機能的に表現された抗体の医薬用途発明に係る特許において、効果を実証した記載が実施例一つのみであっても、実施可能要件及びサポート要件を満たすと判断された事案である。
本件訂正発明1は、具体的な構造を特定しない「抗IL-4R抗体」という機能的な表現を発明特定事項とする医薬用途発明であり、その発明の効果が具体的なデータをもって明細書に記載されていた抗IL-4R抗体は、REGN-668(すなわち、デュピルマブ)のみであった。このたった1つの抗体による効果の実証により、その上位概念である抗IL-4R抗体という広い範囲まで記載要件を充足するのか否かは、本件訴訟の大きな注目点でもあった(判決の内容は前述したとおり)。
サポート要件について判断した有名な「偏光フィルムの製造法」事件大合議判決(2005.11.11知財高裁平成17年(行ケ)10042)が説示したサポート要件に適合する二つの場合について、田村善之先生は、一方の場合を、特許発明に係る技術的思想を演繹的に記載することによってサポート要件を充足させることから「技術的意味型」、他方の場合を、実施例を多数揃えることにより帰納的に記載することによってサポート要件を充足させることから「具体例型」と呼び、そして、このような方策はパラメータ特許に限られるものではないから、一般的な通有性を誇るものと理解できるとし、その後の「被覆硬質部材」事件判決(2008.06.12知財高裁平成19(行ケ)10308)も、この大合議判決を踏襲しつつ、実施例の記載が不十分であるために「具体例型」に当たらない場合、「技術的意味型」として救済されるのは「特許請求の範囲に記載された発明によって課題解決若しくは目的達成等が可能となる因果関係又はメカニズムが、明細書に開示されいているか又は当業者にとって明らかであるなどの場合」であることを明らかにしたものであると指摘する(参照: 田村 善之 「サポート要件と実施可能要件と機能的クレイムの関係に関する一考察(1)―クレイムの全範囲にわたって実施可能とする必要があるのか?―」 知的財産法政策学研究 Vol.67, p101 (2023))。
本件訴訟で裁判所は、IL-4Rに結合しIL-4を遮断する作用を有する抗IL-4R抗体であれば、唯一データが示されている1つの抗体(デュピルマブ)に限らず、本件患者に対して治療効果を示すだろうことを「演繹的に導かれる推論として・・・理解されるもの」であると述べ、発明の課題を解決できるとの認識が得られるとしてサポート要件を充足すると判断した。すなわち、本件は「技術的意味型」としてサポート要件に適合すると判断された事例といえる。
近年、「機能的に表現された抗体」に関するクレームの記載要件を巡る裁判が注目を集めているが、本件訂正発明1は「機能的に表現された抗体」そのものではなく、「機能的に表現された抗体の医薬用途」に関するクレームである点が異なる。では、「機能的に表現された抗体」クレームよりも、「機能的に表現された抗体の医薬用途」クレームの方が記載要件を満たしやすいのだろうか。
「機能的に表現された抗体の医薬用途」クレームは、医薬用途の限定があるため、発明の技術的範囲は一見すると「機能的に表現された抗体」クレームよりも狭くなる。また、こうしたクレームは、サポート要件に関して演繹的に導かれる「技術的意味型」と比較的相性が良い場合が多いと考えられる。しかし、医薬用途発明の技術的思想が高度であったり分類や機序が多様であったりする場合には、演繹的にその内容を導く(裏付ける)ことは難しくなり、要件を満たすハードルが高くなる可能性もある(抗体事例ではないが、例えば、痛みの医薬用途発明について記載要件が問われた事件として2022.08.24 ブログ記事「疼痛治療剤リリカ®(プレガバリン)の後発医薬品の特許権侵害訴訟で控訴審(知財高裁)判決出揃う 全ての「痛み」の用途特許は無効、特許権侵害認めず・・・4つの部で本件訂正が新規事項追加か否かへの向き合い方に違い」参照)。
したがって、「機能的に表現された抗体」と「機能的に表現された抗体の医薬用途」のどちらが記載要件を満たしやすいかについては、一般論として一概に結論を出すことは困難である。
「機能的に表現された抗体」クレームの記載要件について話題となった事例の参考記事:
- 2024.01.16 「ロシュ ダイアグノスティックス v. アボット」 知財高裁令和4年(行ケ)10082 ―「抗原Xに結合する抗体」をさらに「特異的に認識して結合する」特徴で特定する意義―
- 2023.01.26 「リジェネロン v. アムジェン」 知財高裁令和3年(行ケ)10093 ―「参照抗体と競合する」抗体クレームのサポート要件充足性と発明特定事項の意義―(『医薬系特許的判例ブログ年報 2023』 Fubuki著 2024年5月発行, p53-84)
- 【速報】2023.05.18 「Amgen v. Sanofi」 米国最高裁No. 21–757 - Amgenの抗PCSK9抗体特許 実施可能要件非充足を理由に無効としたCAFC判決を米国最高裁も支持 -(『医薬系特許的判例ブログ年報 2023』 Fubuki著 2024年5月発行, p180-182)
- 機能的表現抗体クレイムの終焉か ―Amgen事件米国最高裁判決を受けBaxalta社の抗体特許を無効とする判決(Baxalta v. Genentech CAFC 2022-1461)―(『医薬系特許的判例ブログ年報 2023』 Fubuki著 2024年5月発行, p222-229)
5.おわりに
本稿では、アトピー性皮膚炎患者を対象としたデュピルマブの有効性を確認する臨床試験プロトコルの公開と、臨床試験結果に基づく特許出願の関係性について整理し、本件訴訟における裁判所の判断や欧米の審査状況を紹介した。
その中で、臨床試験プロトコルの公開が特許性に与える影響を踏まえ、臨床試験開始前に出願すべきか、あるいは試験結果が得られてから出願すべきかというジレンマに言及した。
本稿を通じて述べてきたように、臨床試験結果に基づく医薬用途発明の特許出願のタイミングは、発明内容や技術常識に依存するものの、単に臨床試験結果の有無や技術的背景の観点だけで決まるものではない。
各国の特許制度における新規性・進歩性、記載要件、臨床試験プロトコルの公開がもたらす影響、さらには企業のビジネス戦略や市場における競争環境も重要な要素であり、それら観点を総合的に考慮しつつ、ビジネス的観点から最適な出願戦略を構築する必要がある。
そのためには、臨床試験開始時点でのプロトコル公開によるリスクをどのように管理し、どのようなタイミングで出願を行っていくか、どのような内容を明細書に組み込んでいくかについて、知財部門は開発部門と連携して、臨床試験の計画段階から出願戦略を練り上げることが求められる。
早い段階から各部門が連携し、迅速かつ的確な対応が不可欠であり、このような総合的な視点を持ったアプローチが、医薬品開発における成功の鍵となるだろう。
答えのない問いに取り組むのも知財の醍醐味だよ。
AIなら答えをすぐ出してくださいよ。ピポ先輩~。
・・・
アシスタント:

Robot icons created by Freepik – Flaticon; Robot icons created by Freepik – Flaticon: Robot cat icons created by Freepik – Flaticon

コメント